 Newsletter
Newsletter Contacto
Contacto
Dr. Javier Benítez.
Genetista. Expresidente del Patronato y miembro del Comité Científico de la Fundación QUAES
La prevención es uno de los métodos prioritarios de la medicina actual para evitar el desarrollo de enfermedades y/o malformaciones muy severas en la persona. Las técnicas de diagnóstico prenatal que permiten un cribado prácticamente universal de las principales alteraciones cromosómicas es un buen ejemplo, lo mismo que los cribados genéticos que se realizan en los primeros días de vida del recién o las vacunas en la época infantil, en adultos y en las personas mayores. El objetivo es el mismo, evitar el desarrollo de patologías en su mayoría severas que aparecen en las distintas etapas de la vida.
El conocimiento de las bases genéticas de la enfermedad está abriendo una nueva dimensión al permitir identificar y seleccionar a aquellas personas de la población general que van a desarrollar con una alta probabilidad, enfermedades muy diversas que conllevan además un alto coste sanitario; enfermedades neurológicas, o cardiovasculares, o bien oncológicas, es decir las enfermedades comunes de las que fallecen la mayoría de los humanos, son ejemplos donde poder actuar de una manera individualizada con un tratamiento y seguimiento acorde al riesgo que presentan.
Las enfermedades comunes son el resultado de la combinación de múltiples factores genéticos que confieren cada uno de ellos un riesgo muy pequeño (o una protección), y de factores “no genéticos”, propios, sociales, o ambientales que también confieren un pequeño riesgo o protección, de manera que el resultado final de esa interacción multifactorial hará que las personas tengan una mayor o menor probabilidad de desarrollar un cáncer, o un párkinson, o una enfermedad coronaria.
Desde hace tiempo se conocían muchos de esos factores de susceptibilidad, ambientales o propios de cada individuo, pero no así los factores genéticos. El proyecto “Genoma Humano” presentado en el año 2001, puso de manifiesto la existencia de millones de variantes en nuestro genoma que conocemos con el nombre de SNPs (Single Nucleotide Polymorphisms) y que son los responsables de las diferencias entre los humanos. Estos SNPs son en su mayoría polimorfismos que afectan a una sola base, sin ninguna repercusión para el individuo, pero en ocasiones estos SNPs pueden ser auténticas mutaciones genéticas que pueden modificar muy ligeramente o totalmente, la proteína final de ese gen y su función.
Los SNPs explicarían no solo las diferencias fenotípicas entre las poblaciones humanas sino también las diferencias individuales, la mejor o peor respuesta a determinados fármacos, o la mayor o menor susceptibilidad a desarrollar una enfermedad.
¿Cómo se pueden identificar los SNPs de susceptibilidad entre los millones que tenemos en nuestro genoma?
Hace dos décadas esa era la pregunta del millón. Se conocían algunos genes y la función que tenían. Algunos participaban en la reparación de nuestro ADN; otros en controlar el ciclo celular; otros se asociaban con la obesidad, y así sucesivamente. Si se encontraba un polimorfismo (SNP) en un gen asociado con la obesidad y se quería conocer qué efecto podía tener ese SNP, se estudiaba el mismo SNP en un grupo de individuos obesos y en un grupo de individuos de peso normal, lo que se conoce como un estudio caso-control. Se trataba de averiguar si ese SNP se presentaba más frecuentemente en la población obesa, en la normal, o se encontraba en ambas por igual. En el primer caso se podía suponer que la presencia de ese SNP en el gen se asociaba con una tendencia a la obesidad, mientras que, si era más frecuente en la población normal, el SNP sería protector. Si se encontraba en la misma proporción en casos y en controles sería un SNP neutro, uno de los muchos polimorfismos que tenemos.
Los años posteriores nos permitieron asistir a una revolución tecnológica que permitía el estudio de miles o millones de SNPs a la vez, y a la formación de consorcios internacionales que agrupaban a miles de pacientes y de controles de todas partes del mundo permitiendo el estudio de cientos de miles de individuos. De esa manera se empezó a conocer las bases genéticas de muchas enfermedades comunes. Ese conocimiento nos permitió iniciar estudios con el objetivo inicial de poder estratificar a la población general en base al mayor o menor riesgo de desarrollar una enfermedad. Se podían identificar qué SNPs se encontraban sobre representados en una parte de la población (población de riesgo), y darles un valor o peso (odd ratio) (OR), generalmente muy pequeñito, menor de 1.3 por regla general. Pero si esa subpoblación tenía muchos de esos SNPs sobre representados, el OR sería muy elevado, pudiendo ser tan alto como el efecto de un gen responsable de una enfermedad monogénica (la hemofilia, o el corea de Huntington, o la fibrosis quística, o la distrofia muscular de Duchenne entre otros). De manera que un individuo sano, pero portadora de 30 ó 40 de esos SNPs , sería un individuo de alto riesgo para desarrollar la enfermedad objeto de estudio, mientras que otro que solo fuera portador de 3 o 4 de esos SNPs, tendría un riesgo muy pequeño (probabilidad menor del 5%). Estábamos siendo capaces de estratificar dentro de una población general, un pequeño grupo de muy alto riesgo, un pequeño grupo de muy bajo riesgo, y un amplio grupo de riesgo poblacional, a los que se podría tratar de manera diferente. La campana de Gauss al servicio de la medicina.
Una rápida revisión a la situación actual. Algunos ejemplos
En el año 2013 se publicó el primer estudio amplio en cáncer de mama por parte del Consorcio Internacional de Cáncer de Mama ( Michailidou K et al, Nat Genet 353, 2013). Se analizaron 100.000 SNPs en 50.000 mujeres con cáncer y mujeres de la población general procedentes de diferentes países europeos y norteamericanos. Se identificaron 41 SNPs de riesgo que permitieron en posteriores estudios estratificar a sucesivas poblaciones en mujeres de muy alto riesgo (alrededor de un 40% de probabilidades de desarrollar cáncer) y de muy bajo riesgo (menos del 5% de probabilidad) en comparación con el riesgo poblacional, un 12% aproximadamente. Uno de estos estudios se publicó en el año 2020 en población española con un número mayor de SNPs, 94, e incorporando diferentes factores de riesgo como son los antecedentes familiares, edad de menopausia, edad al primer hijo, densidad mamaria y biopsias previas (Triviño JC et al, BMC 2020). Los resultados combinando los datos genéticos y personales, permitieron agrupar a un 20% de las mujeres de la población general como de alto riesgo, con un valor predictivo positivo del 70%, mientras que en el grupo de mujeres con cáncer solo un 2% se hubieran clasificado como de bajo riesgo (valor predictivo negativo 99%) (Fig.1)
Fig.1:La población general la podemos estratificar en población de alto riesgo, moderado riesgo y bajo riesgo a desarrollar cáncer de mama en base a un conjunto de 94 SNPs y una decena de factores de riesgo no genéticos
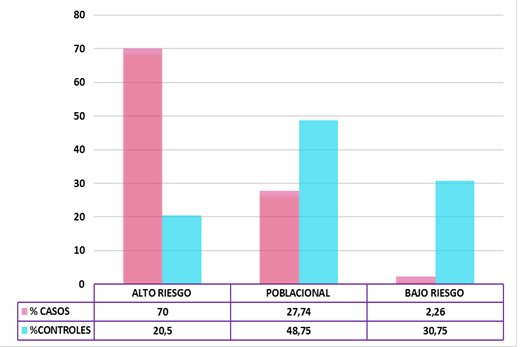
Actualmente hay múltiples publicaciones en diferentes tipos de cáncer, con series de distintas partes del mundo, con un número de SNPs mayor, y con factores modificables (tabaco, alcohol, obesidad), y no modificables (densidad, menopausia, hijos…). Todos ellos con resultados muy similares que ponen de manifiesto la posibilidad de implementar nuevas estrategias preventivas basadas en la medicina individualizada.
La enfermedad de Parkinson, la esquizofrenia, el trastorno bipolar, el autismo, o la depresión mayor, son ejemplos en el ámbito de la neurogenética que han sido estudiado extensamente a pesar de la complejidad y el solapamiento de estas enfermedades. Un solapamiento clínico que se debe en buena medida a compartir varios de estos genes de susceptibilidad. Este hecho es una característica común no solo en patología neurogenética sino en otras enfermedades comunes (cáncer). En los primeros, los SNPs pertenecientes a las vías de señalización, targets de antipsicóticos o los bloqueantes de los canales calcio suelen ser repetitivos, mientras que en cáncer lo son los genes de reparación, de señalización o transmisores de señales. Recientemente se han identificado 36 genes asociados a trastorno bipolar que no solo explican la mayor susceptibilidad a desarrollar la enfermedad, sino que algunos de ellos están también alterados en esclerosis lateral amiotrófica, autismo o enfermedad de Huntington (Nature: 2025 Jan 22. doi: 10.1038/s41586-024-08468-9). En Parkinson los estudios permiten diferenciar subpoblaciones de la población general con un riesgo de 3 a 4 veces superior a la media de desarrollar parkinson, una vez estratificada la población por edad, sexo y factores exógenos y personales de susceptibilidad.
Las enfermedades cardiovasculares constituyen la primera causa de muerte en nuestro País. Se conocen varios factores de riesgo que han permitido elaborar modelos de predicción clínicos usados en muchos países para tener una predicción del riesgo de una persona (SCORE2). Los factores genéticos están completando esos modelos y mostrando el interés que tendría el utilizar la combinación de ambos e incorporarlo a la práctica clínica. En el más reciente de los trabajos para valorar la utilidad de la combinación se han analizado más de 430.000 muestras procedentes del UK Biobank, que recoge factores genéticos (SNPs) y factores personales de riesgo (Li L et al, European Heart Journal (2024) 45, 1843 . Se comprobó que los factores genéticos eran independientes de los factores clínicos, y que, al incorporar esos factores genéticos al perfil de la persona, un 10% de los individuos que se encontraban clasificados como de riesgo poblacional pasaban a incorporarse al subgrupo de alto riesgo, y viceversa, un tercio de las personas de los agrupados como de alto riesgo pasaban a tener un riesgo menor. Estos resultados podrían ser muy importantes desde el punto de vista del tratamiento y seguimiento de los pacientes si se pudieran utilizar en la clínica rutinaria.
¿Por qué no se utilizan actualmente estos modelos predictivos?
A pesar de las ventajas que representan los modelos basados en la integración genética-clínica, su utilización es minoritaria porque se necesitarían resolver algunos problemas. El primero es que se demostrara fehacientemente la capacidad de estratificación que tienen estos modelos y su utilidad clínica. Esto es algo que en determinadas patologías ya está ampliamente demostrado en base a estudios retrospectivos y las curvas de la población sana y la población enferma se separan de manera altamente significativa. Sin embargo, para evaluar la mejora que se obtendría en un seguimiento y tratamiento personalizado en función de la estratificación se requeriría tiempo (años) y eso es algo en lo que se sigue trabajando. El segundo problema se refiere a la interpretación y comunicación de los resultados. La genómica incluye conceptos nuevos y complicados que para un clínico son difíciles de entender y por supuesto de transmitir, y el hecho de hablar de probabilidades agudiza esta situación. La formación genética en esta área sería un requisito imprescindible que la falta de especialidad agudiza. Finalmente, la introducción en las guías clínicas daría el impulso necesario para avanzar en estos aspectos, porque realmente, sin estar en una guía clínica, el primer punto es muy difícil de considerar por las administraciones sanitarias, y el segundo por la academia.
Los estudios pilotos deberían ser una buena solución contando con la cooperación de equipos multidisciplinares que diseñaran de forma rigurosa todos los puntos que deberían cumplir este tipo de estudios. Lo cierto es que poder tener un subgrupo de personas con una alta probabilidad de desarrollar una enfermedad y tener los mecanismos necesarios para evitar ese desarrollo, sería un gran avance.
La Cátedra QUAES-UPF se suma a la 25 edición de la Semana Mundial del Cerebro con la organización del primer Foro Alzheimer …
Del 27 al 31 de marzo se reunieron en la Fundación QUAES neurólogos, pediatras, profesores y psicólogos, junto a profesionales de …

El viernes 17 de junio tuvo lugar la VII Jornada de la Federació Salut Mental CV, que por primera vez se centraba …