 Newsletter
Newsletter Contacto
ContactoEntre enero de 2020 y abril de 2021 se han depositado en la base de datos GISAID más de un millón de secuencias de SARS-CoV-2, el coronavirus responsable de la pandemia de COVID-19. A éstas hay que añadir otras ≈260 000 en Genbank. Aunque la mitad de las secuencias de GISAID son de Reino Unido y Estados Unidos, en total son 172 países los que han aportado secuencias. Esto supone un esfuerzo global sin precedentes, sobre todo teniendo en cuenta que no existe ningún proyecto ni directriz global, sino que se trata de cientos de iniciativas independientes de gobiernos y grupos de investigación de todo el mundo en un movimiento de base (bottom-up) en toda regla. En España hay grupos que han secuenciado por su cuenta y con recursos propios, pero además el Instituto de Salud Carlos III ha financiado un proyecto de alcance nacional, SeqCOVID, que ha producido excelentes resultados
La secuenciación del genoma completo del virus es necesaria para entender la pandemia y su evolución. Los genomas de los virus contienen información genética, que es necesaria para el virus y su ciclo vital, pero que además es útil para los investigadores porque permite diferenciar unos linajes de otros y trazar la evolución del virus.
Desde que empezó la pandemia la población del virus ha generado múltiples linajes que se han dispersado por todo el mundo, cada uno acumulando mutaciones diferentes y divergiendo de la secuencia original. Las secuencias genómicas trazan esa historia (Figura 1).
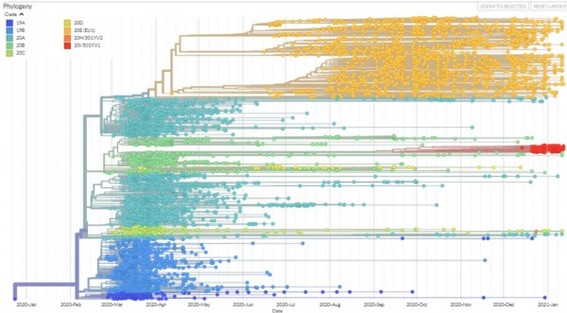
Figura 1: Árbol filogenético de las secuencias españolas de SARS-CoV-2 depositadas en GISAID y obtenido de NextSpain. El árbol tiene el origen a la izquierda. La escala horizontal indica el tiempo desde el inicio de la pandemia y la escala vertical es una escala arbitraria de divergencia evolutiva. En el árbol se diferencia claramente la primera ola de la pandemia (febrero-junio de 2020, en tonos azules, verdes y amarillos), que llegó de manera simultánea por varias vías representadas por diversos clados o linajes que, aunque todavía tenían pocas mutaciones, ya eran claramente diferenciables. La segunda y tercera olas (julio 2020 a enero 2021) estuvieron dominadas por un solo clado, EU1, originado muy posiblemente en España (en color naranja). Hacia el final de la tercera ola llegó la variante 501Y.V1 (también llamada británica, en rojo) que se ha convertido en la mayoritaria en los últimos meses.
Las secuencias muestran también convergencia evolutiva, la aparición de las mismas mutaciones de manera independiente en diferentes linajes señalando la ocurrencia de procesos de selección natural (en respuesta a la presión inmunológica, por ejemplo). Esto va a ser muy importante para seguir la evolución del virus en la era post-vacunal y detectar variantes capaces de escapar a la respuesta inmunológica producida por las vacunas. Por otra parte, con una orientación más clínica, se está estudiando el impacto de las diferentes variantes, sus frecuencias de transmisión y sus patrones de infección. Además, la secuenciación se está utilizando a escala local para caracterizar y delimitar brotes epidémicos y a escala de pacientes individuales para confirmar los casos de reinfección. Y cuando por fin se desarrollen agentes antivirales habrá que monitorizar sus actividades sobre las diferentes variantes y estudiar el posible escape de variantes con mutaciones de que confieran resistencia.
En definitiva, para conocer al virus secuenciamos su genoma.
Fuente: Jesús Mingorance http://www.madrimasd.org/ 25 abril, 2021

Empieza nuestro concurso ¿Qué es para ti el ADN?, con el que tratamos de impulsar el interés por la ciencia entre los …
Os invitamos a escuchar el podcast del programa «A Buenas Horas» de Gestiona Radio, presentado por Miguel Angel Pastor. La intervención del …
En el marco del programa europeo Horizonte 2020 (proyecto ERA-NET) Una investigación impulsada por la Cátedra QUAES-UPF descubre genes que protegen a …